GRN Inference: P22 Mouse Brain¶
STARNet workflows have two main parts: model training, followed by gene regulatory network (GRN) module inference. The inferred GRNs can then be reused for downstream analyses such as spatial trajectory inference, GWAS interpretation, and drug response analysis.
This tutorial uses paired spatial RNA and ATAC data from P22 mouse brain. It walks through STARNet training, GRN inference, module scoring, and spatial visualization. The trained spot and gene embeddings are reused as inputs for GRN inference.
import warnings
import os
warnings.filterwarnings("ignore")
os.environ["PYTHONWARNINGS"] = "ignore::FutureWarning"
import scanpy as sc
import anndata as ad
import STARNet as ST
import pandas as pd
import pickle
Stage 1: Model Training and GRN Inference¶
Read data¶
This tutorial uses the P22 mouse brain dataset. Download the tutorial data from Links to Data Folder, then place the Drive folder next to this notebook so paths such as Drive/Datasets/P22_Mouse/... resolve correctly. You can download only the files used here or the full data folder.
adata_rna = sc.read_h5ad("Drive/Datasets/P22_Mouse/Raw_Data/adata_rna.h5ad")
adata_atac = sc.read_h5ad("Drive/Datasets/P22_Mouse/Raw_Data/adata_atac.h5ad")
Here, we create a ST.model.STARNet object from the paired RNA and ATAC AnnData objects. If a GPU is available, set device='cuda:N' to choose the GPU used for training.
starnet_obj = ST.model.STARNet(adata_rna, adata_atac, device='cuda:3')
Next, preprocess() prepares the paired multi-omics data for model training. This step performs quality control, selects highly variable genes and peaks, normalizes the data, and constructs the required scRNA, scATAC, cell-neighbor, and peak-to-gene graphs.
starnet_obj.preprocess()
==================================================
Step 1: Data Alignment and Initialization
📄 Data Alignment Results:
✓ adata_rna: Dataset shape: 9215 spots × 22914 genes
✓ adata_atac: Dataset shape: 9215 spots × 278227 peaks
Converting adata_rna.X to csr_matrix format.
✅ Conversion complete.
==================================================
Step 2: Processing RNA Data
Filtering genes: min_cells=23
Running RNA Leiden clustering (res=0.2)...
==================================================
Step 3: Processing ATAC Data
Selecting top 40000 ATAC peak features.
Running ATAC Leiden clustering (res=0.2)...
Final RNA shape: (9215, 14484)
Final ATAC shape: (9215, 40000)
==================================================
Step 4: Building Heterogeneous HyperGraph
🧬 RNA graph nodes prepared: 14484 genes
🧩 ATAC graph nodes prepared: 40000 peaks
Building Dual-Modality Spot-Spot matrices...
Using 'leiden' clusters from obs.
Constructed dual-modality matrix for K=3.
Constructed dual-modality matrix for K=4.
Constructed dual-modality matrix for K=8.
✅ Graph data moved to device: cuda:3
We then train STARNet for 600 epochs, which is the recommended setting for these tutorial datasets. Training stores the spot/cell embedding in adata_rna.obs and the gene embedding in adata_rna.uns for later GRN inference.
starnet_obj.train(epochs=600, eval_every=600)
==================================================
🔍 Step 1: STARNet Model Initialization and Configuration
📄 Model Parameters:
✓ Hidden Dim: 128
✓ Output Dim: 128
✓ Device: cuda:3
✓ SSL Num Neg: 10240
Lightning model built with ComplexConv_v4 architecture.
==================================================
🔍 Step 2: Starting STARNet Training Loop
📄 Training Configuration:
✓ Max Epochs : 600
✓ Device : cuda:3
✓ Evaluation Every : 600 epochs
✓ Checkpointing : True
✓ TensorBoard Log Dir : ./lightning_logs/STARNet/version_6
==================================================
📊 Epoch Summary: epoch=1/600, best_total_loss=7.2521
📊 Epoch Summary: epoch=600/600, best_total_loss=0.6032
Evaluating clustering using representation 'cell_embedding'...
==================================================
📊 Training Summary:
✓ Completed Epochs : 600
✓ Best Total Loss : 0.6032
✅ Epoch-level training visualization finished.
==================================================
✅ Training finished!
✓ TensorBoard logs saved to: ./lightning_logs/STARNet/version_6
✓ Checkpoints saved to: ./lightning_logs/checkpoints
The trained RNA AnnData object is shown below and saved for reuse in the GRN inference steps.
print(f'RNA data after training: {starnet_obj.adata_rna}')
starnet_obj.adata_rna.write_h5ad("Drive/Datasets/P22_Mouse/Process_Data/adata_rna_trained.h5ad")
RNA data after training: AnnData object with n_obs × n_vars = 9215 × 14484
obs: 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_50_genes', 'pct_counts_in_top_100_genes', 'pct_counts_in_top_200_genes', 'pct_counts_in_top_500_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt', 'total_counts_ribo', 'log1p_total_counts_ribo', 'pct_counts_ribo', 'total_counts_hb', 'log1p_total_counts_hb', 'pct_counts_hb', 'leiden', 'pre_clusters'
var: 'mt', 'ribo', 'hb', 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts', 'n_cells'
uns: 'log1p', 'pca', 'neighbors', 'leiden', 'umap', 'gene_embedding', 'pre_clusters_colors', 'leiden_colors'
obsm: 'X_spatial', 'X_pca', 'cell_embedding', 'X_umap'
varm: 'PCs'
obsp: 'distances', 'connectivities'
Load multi-omics data¶
For GRN inference, you can either continue with the AnnData object trained above or load the pre-trained dataset provided with the tutorial files.
# Load data from the trained model or from a saved state
# optional
# adata_rna = starnet_obj.adata_rna
# adata_atac = starnet_obj.adata_atac
adata_rna = sc.read_h5ad('Drive/Datasets/P22_Mouse/Process_Data/adata_rna_trained.h5ad')
adata_rna_raw = sc.read_h5ad("Drive/Datasets/P22_Mouse/Raw_Data/adata_rna.h5ad")
adata_atac = sc.read_h5ad("Drive/Datasets/P22_Mouse/Raw_Data/adata_atac.h5ad")
Before GRN inference, we add the raw RNA counts to the count layer because the downstream GRN calculations use raw count values.
# Gene embedding
adata_rna.uns['gene_embedding'] = ad.AnnData(
adata_rna.uns['gene_embedding'],
obs=pd.DataFrame(index=adata_rna.var_names)
)
# Clean cell names
for x in [adata_rna, adata_rna_raw, adata_atac]:
x.obs_names = [i.split('-')[0] for i in x.obs_names]
# Add raw counts
adata_rna.layers['counts'] = adata_rna_raw[adata_rna.obs_names, adata_rna.var_names].X
adata_atac.layers['counts'] = adata_atac.X.copy()
print(f'RNA data info:\n{adata_rna}')
print(f'ATAC data info:\n{adata_atac}')
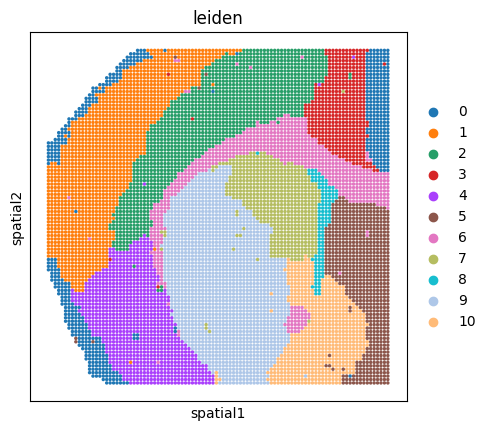
sc.pl.spatial(adata_rna, color='leiden', spot_size=1.25)
RNA data info:
AnnData object with n_obs × n_vars = 9215 × 14484
obs: 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_50_genes', 'pct_counts_in_top_100_genes', 'pct_counts_in_top_200_genes', 'pct_counts_in_top_500_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt', 'total_counts_ribo', 'log1p_total_counts_ribo', 'pct_counts_ribo', 'total_counts_hb', 'log1p_total_counts_hb', 'pct_counts_hb', 'leiden', 'pre_clusters'
var: 'mt', 'ribo', 'hb', 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts', 'n_cells'
uns: 'gene_embedding', 'leiden', 'leiden_colors', 'log1p', 'neighbors', 'pca', 'pre_clusters_colors', 'umap'
obsm: 'X_pca', 'X_spatial', 'X_umap', 'cell_embedding'
varm: 'PCs'
layers: 'counts'
obsp: 'connectivities', 'distances'
ATAC data info:
AnnData object with n_obs × n_vars = 9215 × 278227
obs: 'n_fragment', 'frac_dup', 'frac_mito', 'Sample'
obsm: 'X_spatial'
layers: 'counts'
Load genomic reference files and infer GRNs¶
With the multi-omics data prepared, we load the genomic reference folder and infer GRNs. use_rep selects the embedding used to group similar spots/cells. pvalue_regulatory=0.2 controls the p-value threshold for candidate cis-regulatory links, and moranI_threshold=0.01 keeps transcription factors with Moran’s I scores above the threshold as spatially variable regulators.
genomic_data_pathway = 'Drive/Datasets/Reference/mouse_vM25'
adata_rna = ST.grn.infer_grn_from_multiomics(adata_rna,
adata_atac,
genomic_data_pathway,
use_rep='cell_embedding',
pvalue_regulatory=0.2,
moranI_threshold=0.01,
n_jobs=5)
==================================================
🧬 Starting Spatially Specific GRN Inference Pipeline in STARNet
==================================================
==================================================
🔍 Step 1: Identifying Genomic Reference Files
📄 Target Directory: Drive/Datasets/Reference/mouse_vM25
✅ Genomic files identified and validated.
📄 File Discovery Results:
✓ Motif File : cisBP_mouse.meme
✓ Genome Fasta : gencode_vM25_GRCm38.fa
✓ Annotation (GFF) : gencode_vM25_GRCm38.gff3
✓ Annotation (GTF) : gencode_vM25.chr_patch_hapl_scaff.annotation.gtf
✅ Reference genome and annotation paths successfully loaded.
==================================================
🔍 Step 2: Peak GC Content Analysis
⚙️ Calculating GC proportion for peaks of spatial ATAC-seq data...
==================================================
🔍 Step 3: Spatially Specific Transcription Factor Identification
⚙️ Identifying TFs with significant spatial patterns (Moran's I > 0.01)...
✓ Identify spatially-variable expressed TFs: 135
==================================================
🔍 Step 4: Genomic Data Alignment and Formatting
✅ Genomic data preparation successful.
📄 Processed Stats:
✓ RNA spots/genes : 9215 × 12987
✓ ATAC spots/peaks : 9215 × 278208
✓ Aligned Genes : 12987
==================================================
🔍 Step 5: Primary GRN Construction (Embedding Similarity)
⚙️ Calculating target genes using vectorized cosine similarity...
✓ Total TFs processed : 135
✓ Total Interactions : 67500
✅ Primary GRN construction complete.
==================================================
🔍 Step 6: Metacell Generation
⚠️ CuPy is not installed. Switching to CPU mode. To use GPU acceleration, please install CuPy (https://github.com/cupy/cupy).
⚙️ Calculate the metacells for spatial RNA-seq and spatial ATAC-seq data by using SEACells...
⚙️ Building kernel on cell_embedding...
✓ Generated 122 metacells.
==================================================
🔍 Step 7: GRN Filtering (Stage1)
⚙️ Calculating correlations for 67500 interactions...
📄 Processed Stats:
✓ Threshold : >0.2
✓ Initial TF-target Pairs : 67500
✓ Passed TF-target Pairs : 17018
✓ Removal TF-target Rate : 74.79%
✅ Correlation filtering complete.
⚙️ Mapping peaks to genes (100kb around gene body)...
📄 Processed Stats:
✓ Expand Range : +/- 100,000 bp
✓ Initial Pairs : 17018
✓ Validated Pairs : 16891
✓ Removal Rate : 0.75%
✅ Peak filtering complete.
==================================================
⚙️ Step 8: GRN Filtering using Peak-to-Gene Links (Stage2)
Loading transcripts per gene...
Preparing matrices for gene-peak associations
Computing peak-gene correlations
📄 Processed Stats:
✓ Initial Peak-to-Gene Links : 196,098
✓ Significant Peak-to-Gene Links : 55,310
✓ Removal Rate : 71.79%
✅ Peak-to-Gene filtering complete.
==================================================
🔍 Step 9: Parallel Motif Scanning
⚙️ Identify the motif corresponding to specific transcription factors...
==================================================
📊 Finally Spaitally Specific GRN Inference Results:
==================================================
📊 Final Network Summary:
✓ Total Target Genes : 3,038
✓ Total Regulatory Peaks : 34,669
✓ Total TF-Target Interactions: 11,716
✅ Network added to .uns['grn_df'] and regulatory peaks to .uns['regulatory_peaks']
==================================================
Next, we extract peak-to-gene associations from the GTF gene annotation file in the reference folder. These links connect candidate cis-regulatory elements with nearby genes, allowing STARNet to associate TF binding with spatially specific target gene expression.
gtf_pathway = 'Drive/Datasets/Reference/mouse_vM25/gencode_vM25.chr_patch_hapl_scaff.annotation.gtf'
peak2gene = ST.pp.extract_peak_gene_associations(adata_rna,gtf_file=gtf_pathway)
peak2gene.to_csv('Drive/Datasets/P22_Mouse/Process_Data/peak2gene.links', sep="\t", index=False)
==================================================
🔗 Extracting Peak-Gene Associations for Visualization
==================================================
⚙️ Loading GTF and aligning gene coordinates...
⚙️ Aggregating Peak-to-Gene links from .uns['Peak2Gene']...
⚙️ Parsing genomic coordinates...
⚙️ Calculating scores and formatting...
==================================================
📊 Association Extraction Summary:
✓ Total Raw Links : 55,310
✓ Self-loops Removed : 3
✓ Final Associations : 55,307
✅ Formatting complete.
==================================================
After inferring GRNs and peak-to-gene links, we score each GRN and TF module to quantify how strongly each transcription factor regulates its target genes. These scores support GRN visualization and can also be used as edge weights for downstream cell reprogramming analysis with PriciCE.
# Perform permutation test on all gene regulatory networks
adata_rna = ST.pp.score_all_grn(adata_rna,n_jobs=5)
# Calculate the TF module using various clustering methods and perform cauchy combination tests
adata_rna = ST.pp.score_TF_module(adata_rna,
clustering_method='leiden',
resolution = 2,
groupby='leiden',n_jobs=5)
==================================================
🧬 Calculating GRN Significance via Permutation Test
==================================================
⚙️ Starting parallel scoring for 135 GRNs...
==================================================
✅ Scoring complete! Results stored in:
Full GRN score AnnData: added to .uns['grn']['adata_nlog10_pval']
==================================================
==================================================
🧬 TF Module Analysis: Clustering & Significance
==================================================
⚙️ Preprocessing and running leiden clustering...
✅ Leiden clustering finished: 11 clusters found.
⚙️ Scoring 11 TF modules in parallel...
--------------------------------------------------
✨ All tasks completed successfully!
1. TF Gene Lists: added to .uns['TF_module']['TF_list']
2. Target Gene Lists: added to .uns['TF_module']['target_gene_list']
3. Score AnnData: added to .uns['TF_module']['nlog10_pval_ad']
4. Cauchy Results: added to .uns['TF_module']['cauchy_combination_test']
==================================================
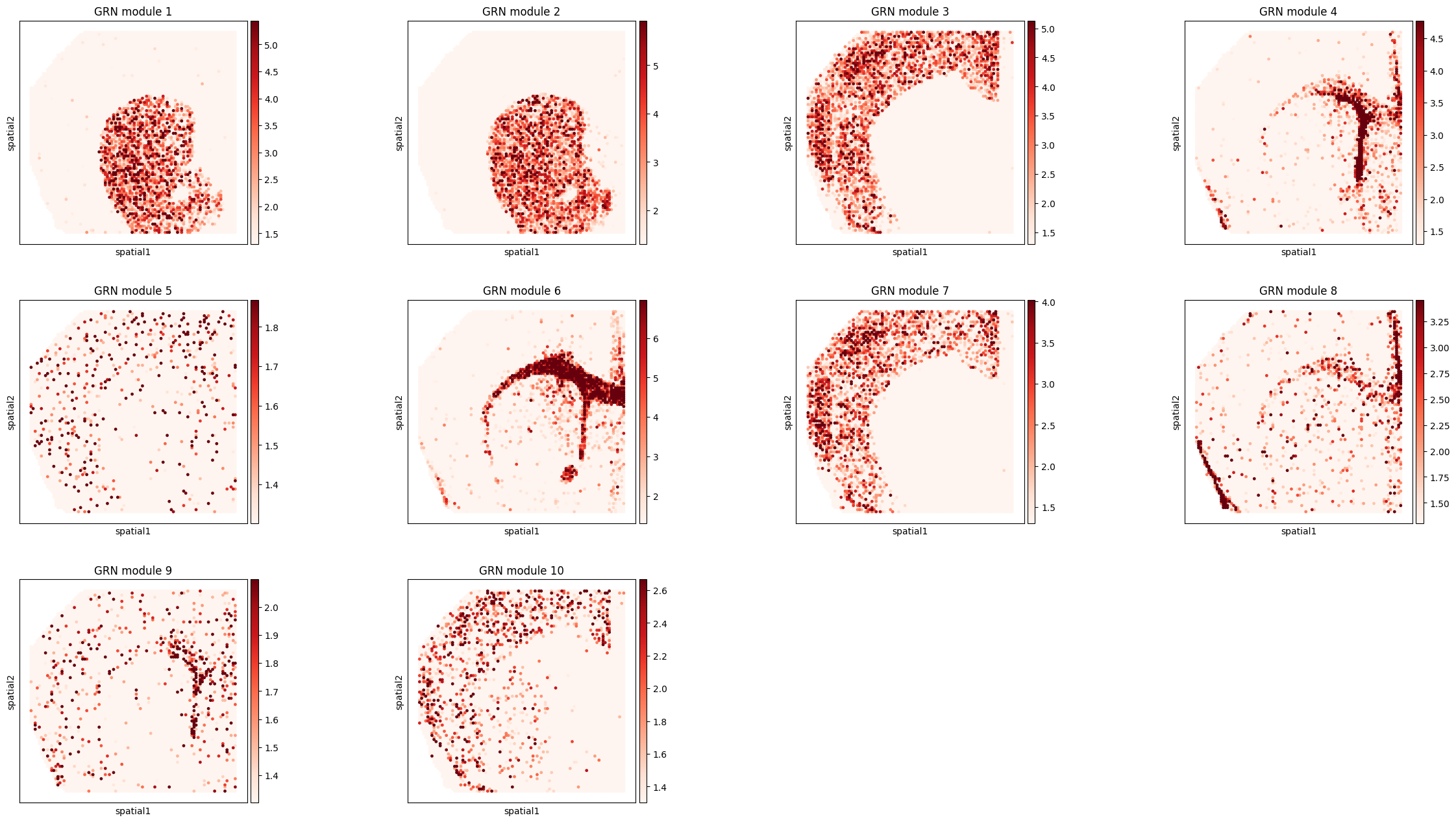
Visualize GRN modules¶
The following cell plots GRN module activity in spatial coordinates. The module patterns should align with known anatomical regions in the P22 mouse brain.
import matplotlib.pyplot as plt
import scanpy as sc
import numpy as np
import anndata as ad
mod_ad = ad.AnnData(adata_rna.uns['TF_module']['nlog10_pval_df'][[str(i) for i in range(10)]])
mod_ad.obsm['spatial'] = adata_rna[mod_ad.obs_names].obsm['spatial']
mod_ad.var_names = [f'GRN module {int(i) + 1}' for i in mod_ad.var_names]
sc.pl.spatial(
mod_ad,
color=mod_ad.var_names,
cmap='Reds',
spot_size=1.5,
vmin=-np.log10(0.05),
vmax='p98',
ncols=4,
)
adata_rna.write_h5ad("Drive/Datasets/P22_Mouse/Process_Data/adata_rna_GRN.h5ad",compression="gzip")